Datapoints#
[1]:
import numpy as np
from rdkit import Chem
from chemprop.data.datapoints import LazyMoleculeDatapoint, MoleculeDatapoint, ReactionDatapoint
Molecule Datapoints#
MoleculeDatapoints are made from target value(s) and either a rdkit.Chem.Mol object or a SMILES.
[2]:
mol = Chem.MolFromInchi("InChI=1S/C2H6/c1-2/h1-2H3")
smi = "CC"
n_targets = 1
y = np.random.rand(n_targets)
[3]:
MoleculeDatapoint(mol, y)
[3]:
MoleculeDatapoint(mol=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d215b0>, y=array([0.30484272]), weight=1.0, gt_mask=None, lt_mask=None, x_d=None, x_phase=None, name=None, V_f=None, E_f=None, V_d=None)
[4]:
MoleculeDatapoint.from_smi(smi, y)
[4]:
MoleculeDatapoint(mol=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d21770>, y=array([0.30484272]), weight=1.0, gt_mask=None, lt_mask=None, x_d=None, x_phase=None, name='CC', V_f=None, E_f=None, V_d=None)
Hydrogens in the graph#
Explicit hydrogens in the graph created by from_smi can be controlled using keep_h and add_h.
[5]:
MoleculeDatapoint.from_smi("[H]CC", y, keep_h=True).mol
[5]:

[6]:
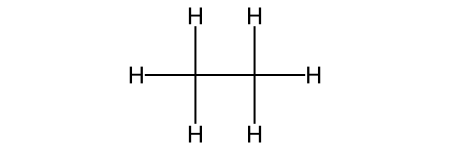
MoleculeDatapoint.from_smi(smi, y, add_h=True).mol
[6]:
Other datapoint properties#
Datapoints can be individually weighted in the loss function.
[7]:
MoleculeDatapoint.from_smi(smi, y, weight=0.5)
[7]:
MoleculeDatapoint(mol=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d21bd0>, y=array([0.30484272]), weight=0.5, gt_mask=None, lt_mask=None, x_d=None, x_phase=None, name='CC', V_f=None, E_f=None, V_d=None)
A string identifier (e.g. a name) can be assigned to a datapoint. If a SMILES is used to make the datapoint, the name defaults to the SMILES, but this can be overwritten.
[8]:
MoleculeDatapoint(mol, y, name="Ethane")
[8]:
MoleculeDatapoint(mol=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d215b0>, y=array([0.30484272]), weight=1.0, gt_mask=None, lt_mask=None, x_d=None, x_phase=None, name='Ethane', V_f=None, E_f=None, V_d=None)
[9]:
MoleculeDatapoint.from_smi(smi, y, name="Ethane")
[9]:
MoleculeDatapoint(mol=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d22180>, y=array([0.30484272]), weight=1.0, gt_mask=None, lt_mask=None, x_d=None, x_phase=None, name='Ethane', V_f=None, E_f=None, V_d=None)
Extra features and descriptors#
Extra datapoint descriptors (like molecule features) will be concatenated to the learned descriptors from message passing and used in the FFN. They are called x_d.
[10]:
n_extra_descriptors = 3
MoleculeDatapoint.from_smi(smi, y, x_d=np.random.rand(n_extra_descriptors))
[10]:
MoleculeDatapoint(mol=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d223b0>, y=array([0.30484272]), weight=1.0, gt_mask=None, lt_mask=None, x_d=array([0.79952846, 0.57058144, 0.61951421]), x_phase=None, name='CC', V_f=None, E_f=None, V_d=None)
Extra atom features, bond features, and atom descriptors are called V_f, E_f, V_d. In this context, features are used before the message passing operations, while descriptors are used after. Extra bond descriptors aren’t currently supported as aggregation ignores the final bond (edge) representations.
[11]:
n_atoms = 2
n_bonds = 1
n_extra_atom_features = 3
n_extra_bond_features = 4
n_extra_atom_descriptors = 5
extra_atom_features = np.random.rand(n_atoms, n_extra_atom_features)
extra_bond_features = np.random.rand(n_bonds, n_extra_bond_features)
extra_atom_descriptors = np.random.rand(n_atoms, n_extra_atom_descriptors)
MoleculeDatapoint.from_smi(
smi, y, V_f=extra_atom_features, E_f=extra_bond_features, V_d=extra_atom_descriptors
)
[11]:
MoleculeDatapoint(mol=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d22420>, y=array([0.30484272]), weight=1.0, gt_mask=None, lt_mask=None, x_d=None, x_phase=None, name='CC', V_f=array([[0.3860953 , 0.64302719, 0.05571153],
[0.06926393, 0.90740897, 0.95685501]]), E_f=array([[0.55393371, 0.29979474, 0.07807503, 0.73485953]]), V_d=array([[0.10712249, 0.33913704, 0.37935725, 0.74724361, 0.49632224],
[0.8496356 , 0.31315312, 0.14000781, 0.58916825, 0.16698837]]))
Reaction Datapoints#
ReactionDatapoints are the same as for molecules expect for:
extra atom features, bond features, and atom descriptors are not supported
both reactant and product
rdkit.Chem.Molobjects or SMILES are required
[12]:
# Keep the atom mapping for hydrogens
rct = Chem.MolFromSmiles("[H:1][C:4]([H:2])([H:3])[F:5]", sanitize=False)
pdt = Chem.MolFromSmiles("[H:1][C:4]([H:2])([H:3]).[F:5]", sanitize=False)
Chem.SanitizeMol(
rct, sanitizeOps=Chem.SanitizeFlags.SANITIZE_ALL ^ Chem.SanitizeFlags.SANITIZE_ADJUSTHS
)
Chem.SanitizeMol(
pdt, sanitizeOps=Chem.SanitizeFlags.SANITIZE_ALL ^ Chem.SanitizeFlags.SANITIZE_ADJUSTHS
)
ReactionDatapoint(rct, pdt, y)
[12]:
ReactionDatapoint(rct=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d22570>, pdt=<rdkit.Chem.rdchem.Mol object at 0x7f64a9d22490>, y=array([0.30484272]), weight=1.0, gt_mask=None, lt_mask=None, x_d=None, x_phase=None, name=None)
The SMILES can either be a single reaction SMILES ‘Reactant>Agent>Product’, or a tuple of reactant and product SMILES. Note that if an Agent is provided, its graph is concatenated to the reactant graph with no edges connecting them.
[13]:
rxn_smi = "[H:1][C:4]([H:2])([H:3])[F:5]>[H:6][O:7][H:8]>[H:1][C:4]([H:2])([H:3]).[F:5]"
from_rxn_smi = ReactionDatapoint.from_smi(rxn_smi, y, keep_h=True)
from_rxn_smi.rct
[13]:

[14]:
rct_smi = "[H:1][C:4]([H:2])([H:3])[F:5]"
pdt_smi = "[H:1][C:4]([H:2])([H:3]).[F:5]"
from_tuple = ReactionDatapoint.from_smi((rct_smi, pdt_smi), y, keep_h=True)
from_tuple.rct
[14]:

Lazy Molecule Datapoints#
LazyMoleculeDatapoints are similar to MoleculeDatapoints, but the rdkit.Chem.Mol object is not computed until the mol property is accessed. This allows for more efficient memory usage when also using cuik-molmaker for featurization. The datapoint stores the featurization options used to create the rdkit.Chem.Mol object. The target(s) y should be provided at the time of initialization by explicitly specifying y=y. If not, it is incorrectly assigned to
_keep_h due to MRO of LazyMoleculeDatapoint.
[ ]:
datapoint = LazyMoleculeDatapoint(
smi, _keep_h=False, _add_h=False, _ignore_stereo=False, _reorder_atoms=False, y=y
) # `Chem.Mol` not yet computed
mol = datapoint.mol # `Chem.Mol` is computed and cached
[ ]:
incorrect_datapoint = LazyMoleculeDatapoint(
smi, y
)
incorrect_datapoint._keep_h